
Для описания изотермы по рис. 2.10а используют уравнения вида:
где К и к -константы.
Приведенные уравнение являются основными в теории мономолеку- лярной адсорбции Ленгмюра. Чаще используется первый вариант, так как в случае адсорбции ПАВ справедливы все уравнения, содержащие величину А , поскольку при этом абсолютная и гиббсовская адсорбции практически одинаковы (А = Г).
При выводе уравнения Ленгмюра физическое взаимодействие на поверхности может быть представлено как квазихимическая реакция:
![]()
где А - адсорбционные центры поверхности; В - распределяемое вещество; АВ - комплекс, образующийся на поверхности.
По мерс увеличения концентрации (давления) вещества В равновесие реакции сдвигается в сторону образования комплекса и свободных центров становится меньше. Константа адсорбционного равновесия по закону действия масс имеет вид:
Введем обозначения:
 [В) = с
; [лв]н=Л и [а= Л 0 , в которых А
- величина адсорбции; А^
- число оставшихся свободными адсорбционных
[В) = с
; [лв]н=Л и [а= Л 0 , в которых А
- величина адсорбции; А^
- число оставшихся свободными адсорбционных
центров, приходящихся на единицу площади поверхности или единицу массы адсорбента. Если А
- величина предельной адсорбции (емкость адсорбционного монослоя), то
![]() Подставляя принятые обозначения в уравнение для константы равновесия, получим выражение для константы
Подставляя принятые обозначения в уравнение для константы равновесия, получим выражение для константы
 , которое после преобразований и дает известное уравнение изотермы мономолекулярной адсорбции Ленгмюра:
, которое после преобразований и дает известное уравнение изотермы мономолекулярной адсорбции Ленгмюра:
![]()
Для газов вместо концентрации используется давление д
(так как концентрация газов и паров при газовой адсорбции практически пропорциональна парциальным давлениям):
![]()
Для характеристики адсорбции используется степень заполнения поверхности . Относительно степени заполнения уравнение (2.9)
можно записать в виде
Константы адсорбционного равновесия в разных видах уравнений Ленгмюра (К , к и к") характеризуют энергию взаимодействия адсорбента и адсорбата: чем сильнее это взаимодействие, тем больше константа адсорбционного равновесия.
Известен другой вариант вывода уравнения Ленгмюра - кинетический, в котором основное внимание уделяется скорости наступления динамичного равновесия процессов адсорбции и десорбции. При этом выводе показывается, что константа адсорбционного равновесия равна отношению констант скорости адсорбции и десорбции:
Для анализа изотермы адсорбции по уравнению Ленгмюра воспроизведем типичную изотерму адсорбции по мономолекулярному механизму (рис. 2.26).

Рис. 2.26.
Анализ изотермы мономолекулярной адсорбции :
При очень малых концентрациях, когда с^О, произведением К с в знаменателе можно пренебречь, поэтому получаем А = А СС -Кс или А = К Г ‘С. Полученные соотношения соответствуют закону Генри и коэффициент пропорциональности К г - константа Генри. По закону Генри
величина адсорбции с ростом концентрации на участке АВ увеличивается линейно;
При больших концентрациях или давлениях, когда произведение К-с» 1, адсорбция стремится к предельному значению А = А #. Это соотношение на участке СД отвечает состоянию насыщения поверхности адсорбента молекулами адсорбата, когда вся поверхность адсорбента покрывается мономолекулярным слоем адсорбата;
в области средних концентраций на участке ВС уравнение Ленгмюра применимо в полной форме.
Физический смысл константы Генри, иногда называемый еще константой распределения, поясняют следующие рассуждения. Если поверхностный слой рассматривать как отдельную фазу, то перераспределение вещества между поверхностным слоем и объемом фазы будет происходить до тех пор, пока химические потенциалы обеих фаз не станут равными:
где p s - химический потенциал в поверхностном слое; p v - химический потенциал объемной фазы.
Учитывая, что , для равновесного состояния имеем , откуда
Если в области малых концентраций активности считать равными концентрациям, то поверхностная концентрация равна адсорбции
a s
= с s
= А
и тогда
![]() Из представленных соотношений и полу
Из представленных соотношений и полу
чается уравнение Генри: А = Кр-с.
Можно получить подобное выражение через давление, учитывая, что в области малых концентраций газ подчиняется закону состояния идеального газа pV = nRT
, откуда
![]() . Подставляя послед
. Подставляя послед
нее соотношение в уравнение адсорбции, получаем:

Уравнения Генри просты по виду, но иногда их бывает вполне достаточно для практических расчетов. На твердых поверхностях область действия этого закона мала из-за неоднородности поверхности. Но даже на однородной поверхности обнаруживается отклонение от линейной зависимости при увеличении концентрации (давления). Это объясняется уменьшением доли свободной поверхности, приводящим к замедлению роста адсорбции.
Отклонения от закона Генри учитывает эмпирическое уравнение, установленное Фрейндлихом и Бедекером на основе изучения адсорбции газов на твердых адсорбентах. Позднее это уравнение было теоретически обосновано Зельдовичем и оказалось применимым и для растворов.
Теория мономолекулярной адсорбции была создана Ленгмюром при изучении адсорбции газов на твердых поверхностях. Основные положения теории состоят в следующем:
- - на поверхности твердого адсорбента имеются активные центры, все они энергетически однородны (поверхность эквипотенциальна) и их количество на единице площади постоянно для данного адсорбента;
- - каждый активный ценгр удерживает только одну молекулу адсорбата, которая закреплена с ним силами физической природы (адсорбция обратима). Адсорбированная молекула образует с центром прочный комплекс и не способна перемещаться по поверхности;
- - учитываются только силы взаимодействия молекулы с адсорбционным центром (без учета взаимодействия между молекулами адсорбага).
Несмотря на жесткие ограничения, теория широко используется и даст хорошую сходимость с практическими результатами для большого количества видов адсорбции. В настоящее время она распространяется на адсорбцию на других границах раздела.
Теория Ленгмюра объясняет адсорбцию ПАВ на границе вода - воздух, когда полярная группа, обладая большим сродством к полярной фазе, втягивается в воду, в то время как неполярный радикал выталкивается в неполярную фазу (воздух) и при малых концентрациях углеводородные цепи «плавают» на поверхности воды (это возможно из-за их гибкости). С ростом концентрации цепи поднимаются и в насыщенном адсорбционном слое занимают вертикальное положение, при этом поверхность воды сплошь покрыта «частоколом» из вертикально ориентированных молекул ПАВ. Значение поверхностного натяжения в этом случае приближается к значению чистого жидкого ПАВ на границе с воздухом. Максимальная адсорбция Г а0 именно поэтому не зависит от длины углеводородного радикала, а определяется только размерами поперечного сечения молекул.
Существование насыщенных адсорбционных слоев позволяет определять размеры молекул ПАВ. Впервые в истории химии размеры молекул были определены именно коллоидно-химическим путем и уже позже подтверждены другими методами. Поскольку в насыщенном слое молекулы плотно упакованы и имеют вертикальную ориентацию, то можно рассчитать важные характеристики мономолекулярного слоя:
Размер поперечного сечения молекул, то есть площадь, занимаемую одной молекулой ПАВ в поверхностном слое («посадочную площадку»):
Длину молекулы ПАВ, равную толщине адсорбционного слоя:
где N А - число Авогадро, р и М - плотность и молекулярная масса поверхностно-активного вещества.
Для определения постоянных параметров проводят преобразование уравнения Ленгмюра к уравнению прямой линии
Представляя экспериментальные данные в обратных осях или в осях , в первом случае по отрезку, отсекаемому на оси ординат при , определяют величину . Тангенс угла наклона прямой позволяет определить отношение и рассчитать значение пре
дельной адсорбции, по которому можно вычислить адсорбционную константу К . Во втором случае, наоборот, отрезок на ординате связан с величиной обратной предельной адсорбции , а по тангенсу угла наклона
Рассмотрим вариант определения констант уравнения Ленгмюра на примере адсорбции в системе вода - изоамиловый спирт. В таблице представлены экспериментальные данные но величинам поверхностного натяжения о растворов различной концентрации с :
Температура опыта составляет 296 К, при которой поверхностное натяжение воды равно 72,28 мДж/м
Будем применять прием графического дифференцирования, для этого построим изотерму поверхностного натяжения
и рассчитаем величины адсорбции по уравнению Гиббса:
![]()
Для упрощения расчетов обозначим через Z величину , тогда ад
сорбция определится выражением

Рис. 2.27.
Величине Z
соответствует отрезок, отсекаемый на ординате касательной и горизонталью, проведенными к точке, соответствующей искомой концентрации. Для примера показано нахождение значения величины Z для точки, соответствующей концентрации 0,125 кмоль/м 3 . В примере значение величины Z равно 3,9 мДж/м 2 . Остальные результаты представлены в табл. 2.3. После этого вычисляем обратные значения концентраций и адсорбций, необходимые для работы с уравнением Ленгмюра в линейном виде:
![]()
Таблица 2.3
Обработка экспериментальных данных_
Продолжение табл. 2.3
На рис. 2.28 построен график в «обратных» осях, найти по нему константы уравнению Ленгмюра К и Г " просто, однако еще проще сделать
это с помощью Excel.
В этом случае записываем уравнение зависимости
 , из которого
, из которого
 (по графику это от
(по графику это от
резок, отсекаемый на ординате при ). Тогда значение предельной составляет Г оо = 2,098 10" 6 моль/м 2 . Это одна из констант уравнения Ленгмюра.
Вторая константа находится из коэффициента перед обратной концентрацией, равного 15500, то есть
 . При известном значе
. При известном значе
Размерность адсорбционной константы = м 3 /кмоль.

Рис. 2.28.
Запишем окончательно уравнение адсорбции с найденными константами:
Подчеркнем правомерность приравнивание величин избыточной гиббсовской и абсолютной адсорбции, так как теория Ленгмюра распространяется на все поверхности (жидкие и твердые), заполняемые по моно- молекулярному механизму.
По полученным результатам построить изотерму адсорбции в рассматриваемом примере можно двумя путями, подставляя в полученное уравнение концентрации или напрямую строя график по данным первой и третьей колонки табл. 2.3 (рис. 2.29).

Рис. 2.29.
Это является наглядной проверкой правильности проведенных расчетов. Полученное с помошью Excel уравнение имеет величину достоверности аппроксимации, равную 0,99. При нанесении на 1рафик точек, для которых адсорбция рассчитывается по уравнению, обнаруживаются небольшие отклонения по сравнению с расположением точек, для которых адсорбция определяется графическом дифференцированием (из касательных). Это связано с близостью значений предельной адсорбции (2,098-10 6 моль/м 2) и адсорбции при концентрации 0,5 кмоль/м 3 (2,073-10 6 моль/м 2), а так же (в меньшей степени) округлениями при проведении расчетов.
При построении графиков вручную надо обращать внимание на такие практические особенности, как усреднение данных. Линию изотермы нужно проводить плавно, располагая между точками, а не отдельными прямыми линиями между соседними точками (рис. 2.30).

Рис. 2.30.
На рис. 2.30 показано семейство касательных при обработке изотермы адсорбции олеата натрия вручную (на оси ординат поверхностное натяжение с размерностью мДж/м 2).
Изотерма адсорбции. Уравнение Фрейндлиха.
Величина адсорбции (абсолютная А или избыточная Г) в каждом конкретном случае определяется температурой Т и давлением р (при газообразном адсорбтиве) или температурой Т и концентрацией С (при адсорбции из растворов). Как правило, в теории адсорбции при рассмотрении адсорбционного равновесия один из этих параметров поддерживается постоянным. Так, уравнение вида А = f (р) Т или Г = f (c) Т, связывающее величину адсорбции с давлением или концентрацией при постоянной температуре, называется изотермой адсорбции. Адсорбция (если она выражена не как избыток, а как полное содержание) всегда возрастает с повышением равновесного давления или концентрации. Так как адсорбция - процесс экзотермический, то при повышении температуры величина адсорбции снижается. На рис. 26.9 приведены основные виды кривых адсорбционного равновесия. Изотермам адсорбции при трех температурах (Т 1 > Т 2 >Т 3) соответствует рис. 26.9а.
Рис 26.9. Кривые адсорбционного равновесия: изотермы (а), изобары (б) и изостеры (в) адсорбции
Уравнение, связывающее величину адсорбции с температурой при постоянном равновесном давлении А = f(T) p или постоянной равновесной концентрации Г = f (Т) с, носит название, соответственно, изобары или изопикны адсорбции (рис. 26.9 -б) ; здесь р 1 > р 2 > р 3 . Уравнение вида р = f (Т) А , изостера адсорбции (рис. 26.9 -в), связывает равновесное давление с температурой при постоянном адсорбированном количестве; в этом случае А 1 >А 2 >А 3 .
Задача любой адсорбционной теории - на базе определенной модели процесса адсорбции составить ее математическое описание. В идеале уравнение должно описывать зависимость равновесной величины адсорбции от концентрации адсорбата в объемной фазе при различных температурах, а также прогнозировать изменение теплоты адсорбции от заполнения адсорбента. Наиболее часто при этом находят уравнение изотермы адсорбции. Форма изотермы адсорбции на твердых телах зависит от многих параметров: свойств адсорбента и адсорбата, взаимодействия адсорбент адсорбат, взаимодействия молекул адсорбата между собой в газовой фазе и в адсорбированном состоянии. В области малыхдавлений (или концентраций) и соответствующих им малых заполнений поверхности взаимодействие между молекулами адсорбата незначительно и зависимость А = f(p) T сводится к простейшей форме, называемой законом Генри :
А = kp или А = k"c (26.20)
где k и к" - адсорбционный коэффициент (или коэффициент Генри), с - концентрация адсорбента в объемной фазе, р - давление пара адсорбата. Коэффициент Генри k является мерой интенсивности адсорбции. Можно показать, что любая теоретическая изотерма должна в пределе (при малых заполнениях) переходить в уравнение Генри.
В области средних концентраций зависимость адсорбции растворенных веществ от концентрации хорошо описывается эмпирическим уравнением Фрейндлиха :
![]() (26.21)
(26.21)
где Х - количество адсорбированного вещества, m - масса адсорбента, βи п - константы, характерные для каждой адсорбционной системы, причем 0 < 1/n < 1 . По Фрейндллиху, n не зависит от заполнения, хотя это утверждение не вполне точно. Этим эмпирическим уравнением часто пользуются для ориентировочных расчетов адсорбции. Чаще всего оно применяется в логарифмической форме:
позволяющей построить линейную зависимость ln А - ln c и графически определить оба постоянных параметра β и n.
Поверхностные явления и адсорбция. Типы адсорбционных взаимодействий. Изотермы адсорбции газов. Уравнение Генри и Лэнгмюра. Полимолекулярная адсорбция, теория БЭТ.
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ И АДСОРБЦИЯ
Поверхностная энергия. Адсорбция
До сих пор свойства гетерогенных систем описывались с помощью параметров и функций состояния, характеризующих каждую из фаз в целом. Однако свойства участка фазы, примыкающего к её поверхности, отличаются от свойств фазы в объеме: фактически частицы, находящиеся на поверхности каждой фазы, образуют особую поверхностную фазу, свойства которой существенно отличаются от свойств внутренних областей фазы. Частицы, расположенные на поверхности, находятся в другом окружении по сравнению с частицами, находящимися в объеме фазы, т.е. взаимодействуют как с однородными частицами, так и с частицами другого рода. Следствием этого является то, что средняя энергия g s частицы, находящейся на поверхности раздела фаз, отличается от средней энергии такой же частицы в объеме фазы g v (причем энергия частицы на поверхности может быть как больше, так и меньше энергии частицы в объеме). Поэтому важнейшей характеристикой поверхностной фазы является поверхностная энергия G s – разность средней энергии частицы, находящейся на поверхности, и частицы, находящейся в объеме фазы, умноженная на число частиц на поверхности N:
![]() (26.1)
(26.1)
Очевидно, что общая величина поверхностной энергии фазы будет определяться величиной её поверхности S. Поэтому для характеристики поверхности раздела, отделяющей данную фазу от другой, вводится понятие поверхностное натяжение σ – отношение поверхностной энергии к площади поверхности раздела фаз; величина поверхностного натяжения зависит только от природы обеих фаз. Как и поверхностная энергия фазы, поверхностное натяжение может иметь как положительное, так и отрицательное значение. Поверхностное натяжение положительно, если находящиеся на поверхности частицы взаимодействуют с частицами этой же фазы сильнее, чем с частицами другой фазы (и, следовательно, g s > g v). Согласно принципу минимума свободной энергии, любая фаза будет стремиться самопроизвольно уменьшить свою поверхностную энергию; поэтому в случае положительного поверхностного натяжения (σ > 0) фаза стремится уменьшить свою поверхность. В случае если σ < 0, поверхностная энергия фазы будет уменьшаться при увеличении площади поверхности.
Влияние поверхностного слоя фазы на её общие свойства определяется долей частиц, находящихся на поверхности, от общего числа составляющих данную фазу частиц, т.е. величиной удельной поверхности фазы S/V (поверхности, приходящейся на единицу объема). Свободную энергию фазы G можно представить как сумму поверхностной G s и объемной G v энергий, пропорциональных соответственно площади поверхности и объему фазы:
Разделив это выражение на объем фазы, получаем:
Из уравнения (IV.4) следует, что при одном и том же количестве фазы (т.е. неизменном объеме) вклад поверхностной энергии в общую энергию фазы возрастает с увеличением удельной поверхности или, иначе говоря, степени дисперсности (раздробленности) фазы. В случае, когда степень дисперсности фазы невелика (удельная поверхность незначительна), вкладом поверхностной энергии в полную энергию фазы обычно пренебрегают. Вклад поверхностного слоя в свойства фазы и системы в целом учитывают при изучении дисперсных систем – гетерогенных систем, одна из фаз которой является сплошной (дисперсионная среда ), а другая – раздробленной (дисперсная фаза ).
На границе конденсированной (т.е. твердой или жидкой) фазы с газом поверхностное натяжение всегда положительно, поскольку частицы конденсированной фазы взаимодействуют друг с другом сильнее, чем с молекулами газа. Согласно принципу минимума свободной энергии, конденсированная фаза будет стремиться самопроизвольно уменьшить свою поверхностную энергию. Это может быть результатом либо уменьшения площади поверхности фазы (именно поэтому капля жидкости в невесомости принимает форму сферы), либо уменьшения поверхностного натяжения при появлении на поверхности раздела фаз новых частиц – молекул газа либо растворенного вещества. Процесс самопроизвольного изменения концентрации какого-либо вещества у поверхности раздела двух фаз называется адсорбцией . Адсорбентом называется вещество, на поверхности которого происходит изменение концентрации другого вещества – адсорбата .
Адсорбция на границе раствор – пар
В жидких растворах поверхностное натяжение σ является функцией от концентрации растворенного вещества. На рис. 4.1 представлены три возможных зависимости поверхностного натяжения от концентрации раствора (т.н. изотермы поверхностного натяжения). Вещества, добавление которых к растворителю уменьшает поверхностное натяжение, называют поверхностно-активными
(ПАВ), вещества, добавление которых увеличивает или не изменяет поверхностное натяжение – поверхностно-инактивными
(ПИАВ).

Рис. 26.1 Изотермы поверхностного Рис. 26.2 Изотерма адсорбции
натяжения растворов ПАВ (1, 2) и ПИАВ. ПАВ на границе раствор – пар
ПИАВ (3)
Уменьшение поверхностного натяжения и, следовательно, поверхностной энергии происходит в результате адсорбции ПАВ на поверхности раздела жидкость – пар, т.е. того, что концентрация поверхностно-активного вещества в поверхностном слое раствора оказывается больше, чем в глубине раствора.
Количественной мерой адсорбции на границе раствор-пар является поверхностный избыток Г (гамма), равный числу молей растворенного вещества в поверхностном слое. Количественное соотношение между адсорбцией (поверхностным избытком) растворенного вещества и изменением поверхностного натяжения раствора с ростом концентрации раствора определяет изотерма адсорбции Гиббса :
График изотермы адсорбции ПАВ представлен на рис. 26.2. Из уравнения (26.5) следует, что направление процесса – концентрирование вещества в поверхностном слое или, наоборот, нахождение его в объеме жидкой фазы – определяется знаком производной dσ/dС. Отрицательная величина данной производной соответствует накоплению вещества в поверхностном слое (Г > 0), положительная – меньшей концентрации вещества в поверхностном слое по сравнению с его концентрацией в объеме раствора.
Величину g = –dσ/dС называют также поверхностной активностью растворенного вещества. Поверхностную активность ПАВ при некоторой концентрации С 1 определяют графически, проводя касательную к изотерме поверхностного натяжения в точке С = С 1 ; при этом поверхностная активность численно равна тангенсу угла наклона касательной к оси концентраций:
Нетрудно заметить, что с ростом концентрации поверхностная активность ПАВ уменьшается. Поэтому поверхностную активность вещества обычно определяют при бесконечно малой концентрации раствора; в этом случае её величина, обозначаемая g о, зависит только от природы ПАВ и растворителя. Исследуя поверхностное натяжение водных растворов органических веществ, Траубе и Дюкло установили для гомологических рядов поверхностно-активных веществ следующее эмпирическое правило:
В любом гомологическом ряду при малых концентрациях удлинение углеродной цепи на одну группу СН 2 увеличивает поверхностную активность в 3 – 3.5 раза.
Для водных растворов жирных кислот зависимость поверхностного натяжения от концентрации описывается эмпирическим уравнением Шишковского :
Здесь b и K – эмпирические постоянные, причём значение b одинаково для всего гомологического ряда, а величина К увеличивается для каждого последующего члена ряда в 3 – 3,5 раза.

Рис. 26.3 Предельная ориентация молекул ПАВ в поверхностном слое
Молекулы большинства ПАВ обладают дифильным строением, т.е. содержат как полярную группу, так и неполярный углеводородный радикал. Расположение таких молекул в поверхностном слое энергетически наиболее выгодно при условии ориентации молекул полярной группой к полярной фазе (полярной жидкости), а неполярной – к неполярной фазе (газу или неполярной жидкости). При малой концентрации раствора тепловое движение нарушает ориентацию молекул ПАВ; при повышении концентрации происходит насыщение адсорбционного слоя и на поверхности раздела фаз образуется слой "вертикально" ориентированных молекул ПАВ (рис. 26.3). Образование такого мономолекулярного слоя соответствует минимальной величине поверхностного натяжения раствора ПАВ и максимальному значению адсорбции Г (рис. 26.1-26.2); при дальнейшем увеличении концентрации ПАВ в растворе поверхностное натяжение и адсорбция не изменяются.
Адсорбция на границе твердое тело – газ
При адсорбции газов на твердых телах описание взаимодействия молекул адсорбата и адсорбента представляет собой весьма сложную задачу, поскольку характер их взаимодействия, определяющий характер адсорбции, может быть различным. Поэтому обычно задачу упрощают, рассматривая два крайних случая, когда адсорбция вызывается физическими или химическими силами – соответственно физическую и химическую адсорбцию.
Физическая адсорбция возникает за счет ван-дер-ваальсовых взаимодействий. Она характеризуется обратимостью и уменьшением адсорбции при повышении температуры, т.е. экзотермичностью, причем тепловой эффект физической адсорбции обычно близок к теплоте сжижения адсорбата (10 – 80 кДж/моль). Таковой является, например, адсорбция инертных газов на угле.
Химическая адсорбция (хемосорбция) осуществляется путем химического взаимодействия молекул адсорбента и адсорбата. Хемосорбция обычно необратима; химическая адсорбция, в отличие от физической, является локализованной, т.е. молекулы адсорбата не могут перемещаться по поверхности адсорбента. Так как хемосорбция является химическим процессом, требующим энергии активации порядка 40 – 120 кДж/моль, повышение температуры способствует её протеканию. Примером химической адсорбции является адсорбция кислорода на вольфраме или серебре при высоких температурах.
Следует подчеркнуть, что явления физической и химической адсорбции чётко различаются в очень редких случаях. Обычно осуществляются промежуточные варианты, когда основная масса адсорбированного вещества связывается сравнительно слабо и лишь небольшая часть – прочно. Например, кислород на металлах или водород на никеле при низких температурах адсорбируются по законам физической адсорбции, но при повышении температуры начинает протекать химическая адсорбция. При повышении температуры увеличение химической адсорбции с некоторой температуры начинает перекрывать падение физической адсорбции, поэтому температурная зависимость адсорбции в этом случае имеет четко выраженный минимум (рис. 26.4).

Рис. 26.4 Зависимость объема адсорбированного никелем водорода от температуры
При постоянной температуре количество адсорбированного вещества зависит только от равновесных давления либо концентрации адсорбата; уравнение, связывающее эти величины, называется изотермой адсорбции.
Теории адсорбции
Единой теории, которая достаточно корректно описывала бы все виды адсорбции на разных поверхностях раздела фаз, не имеется; рассмотрим поэтому некоторые наиболее распространенные теории адсорбции, описывающие отдельные виды адсорбции на поверхности раздела твердое тело – газ или твердое тело – раствор.
Теория мономолекулярной адсорбции Ленгмюра
Теория мономолекулярной адсорбции, которую разработал американский химик И. Ленгмюр, основывается на следующих положениях.
1) Адсорбция является локализованной и вызывается силами, близкими к химическим.
2) Адсорбция происходит не на всей поверхности адсорбента, а на активных центрах , которыми являются выступы либо впадины на поверхности адсорбента, характеризующиеся наличием т.н. свободных валентностей. Активные центры считаются независимыми (т.е. один активный центр не влияет на адсорбционную способность других), и тождественными.
3) Каждый активный центр способен взаимодействовать только с одной молекулой адсорбата ; в результате на поверхности может образоваться только один слой адсорбированных молекул.
4) Процесс адсорбции является обратимым и равновесным
– адсорбированная молекула удерживается активным центром некоторое время, после чего десорбируется; т.о., через некоторое время между процессами адсорбции и десорбции устанавливается динамическое равновесие.

Рис. 26.5 Изотерма мономолекулярной адсорбции
В состоянии равновесия скорость адсорбции равна скорости десорбции. Скорость десорбции прямо пропорциональна доле занятых активных центров (х), а скорость адсорбции прямо пропорциональна произведению концентрации адсорбата на долю свободных активных центров (1 – х):
![]() (26.9)
(26.9)
Отсюда находим х:
Разделив числитель и знаменатель правой части уравнения (26.10) на k A , получим:
 (26.11)
(26.11)
Максимально возможная величина адсорбции Г о достигается при условии, что все активные центры заняты молекулами адсорбата, т.е. х = 1. Отсюда следует, что х = Г / Г о. Подставив это в уравнение (26.11), получаем:
Уравнение (26.13) есть изотерма мономолекулярной адсорбции , связывающая величину адсорбции Г с концентрацией адсорбата С. Здесь b – некоторая постоянная для данной пары адсорбент-адсорбат величина (отношение констант скоростей десорбции и адсорбции), численно равная концентрации адсорбата, при которой занята половина активных центров. График изотермы адсорбции Ленгмюра приведен на рис. 26.5. Константу b можно определить графически, проведя касательную к изотерме адсорбции в точке С = 0.
При описании процесса адсорбции газов в уравнении (26.13) концентрация может быть заменена пропорциональной величиной парциального давления газа:
Теория мономолекулярной адсорбции Ленгмюра применима для описания некоторых процессов адсорбции газов и растворенных веществ при небольших давлениях (концентрациях) адсорбата.
Теория полимолекулярной адсорбции Поляни
На практике часто (особенно при адсорбции паров) встречаются т.н. S-образные изотермы адсорбции (рис. 4.6), форма которых свидетельствует о возможном, начиная с некоторой величины давления, взаимодействии адсорбированных молекул с адсорбатом.

Рис. 26.6 Изотерма полимолекулярной адсорбции
Для описания таких изотерм адсорбции М. Поляни предложил теорию полимолекулярной адсорбции , основанную на следующих основных положениях:
1. Адсорбция вызвана чисто физическими силами .
2. Поверхность адсорбента однородна , т.е. на ней нет активных центров; адсорбционные силы образуют непрерывное силовое поле вблизи поверхности адсорбента.
3. Адсорбционные силы действуют на расстоянии, большем размера молекулы адсорбата. Иначе говоря, у поверхности адсорбента существует некоторый адсорбционный объём , который при адсорбции заполняется молекулами адсорбата.
4. Притяжение молекулы адсорбата поверхностью адсорбента не зависит от наличия в адсорбционном объеме других молекул, вследствие чего возможна полимолекулярная адсорбция.
5. Адсорбционные силы не зависят от температуры и, следовательно, с изменением температуры адсорбционный объем не меняется.
Уравнение Фрейндлиха
Теоретические представления, развитые Ленгмюром и Поляни, в значительной степени идеализируют и упрощают истинную картину адсорбции. На самом деле поверхность адсорбента неоднородна, между адсорбированными частицами имеет место взаимодействие, активные центры не являются полностью независимыми друг от друга и т.д. Все это усложняет вид уравнения изотермы. Г. Фрейндлих показал, что при постоянной температуре число молей адсорбированного газа или растворенного вещества, приходящееся на единицу массы адсорбента (т.н. удельная адсорбция x/m), пропорционально равновесному давлению (для газа) или равновесной концентрации (для веществ, адсорбируемых из раствора) адсорбента, возведенным в некоторую степень, которая всегда меньше единицы:
Адсорбция на границе твердое тело – раствор
Молекулярная адсорбция из растворов
Изотермы адсорбции растворенных веществ из раствора по своему виду аналогичны изотермам адсорбции для газов; для разбавленных растворов эти изотермы хорошо описываются уравнениями Фрейндлиха или Лэнгмюра, если в них подставить равновесную концентрацию растворенного вещества в растворе. Однако адсорбция из растворов является значительно более сложным явлением по сравнению с газовой, поскольку одновременно с адсорбцией растворенного вещества часто происходит и адсорбция растворителя.

Рис. 26.8 Ориентация молекул ПАВ на поверхности адсорбента
Зависимость адсорбции от строения молекул адсорбата очень сложна, и вывести какие-либо закономерности довольно трудно. Молекулы многих органических веществ состоят из полярной (гидрофильной) и неполярной (гидрофобной) группировок, т.е. являются поверхностно-активными веществами. Молекулы ПАВ при адсорбции на твердом адсорбенте ориентируются на его поверхности таким образом, чтобы полярная часть молекулы была обращена к полярной фазе, а неполярная – к неполярной. Так, при адсорбции алифатических карбоновых кислот из водных растворов на неполярном адсорбенте – активированном угле – молекулы ориентируются углеводородными радикалами к адсорбенту; при адсорбции из бензола (неполярный растворитель) на полярном адсорбенте – силикагеле – ориентация молекул кислоты будет обратной (рис. 4.8).
Адсорбция из растворов электролитов
Адсорбция из водных растворов электролитов происходит, как правило, таким образом, что на твердом адсорбента из раствора адсорбируются преимущественно ионы одного вида. Преимущественная адсорбция из раствора или аниона, или катиона определяется природой адсорбента и ионов. Механизм адсорбции ионов из растворов электролитов может быть различным; выделяют обменную и специфическую адсорбцию ионов.
Обменная адсорбция представляет собой процесс обмена ионов между раствором и твердой фазой, при котором твердая фаза поглощает из раствора ионы какого-либо знака (катионы либо анионы) и вместо них выделяет в раствор эквивалентное число других ионов того же знака. Обменная адсорбция всегда специфична, т.е. для данного адсорбента к обмену способны только определенные ионы; обменная адсорбция обычно необратима.
При специфической адсорбции адсорбция на поверхности твердой фазы ионов какого-либо вида не сопровождается выделением в раствор эквивалентного числа других ионов того же знака; твердая фаза при этом приобретает электрический заряд. Это приводит к тому, что вблизи поверхности под действием сил электростатического притяжения группируется эквивалентное число ионов с противоположным зарядом, т.е. образуется двойной электрический слой. Взаимодействие концентрирующихся на поверхности зарядов приводит к понижению поверхностной энергии системы. Для случая специфической адсорбции электролита Песковым и Фаянсом было сформулировано следующее эмпирическое правило (правило Пескова-Фаянса ):
На поверхности кристаллического твердого тела из раствора электролита специфически адсорбируется ион, который способен достраивать его кристаллическую решетку или может образовывать с одним из ионов, входящим в состав кристалла, малорастворимое соединение.
В случае взаимодействия двух атомов:
U – энергия взаимодействия;
U = U ПРИТЯЖ. + U ОТТАЛК.
 - уравнение
Леннарда-Джонса
,
c,
b,
m
= const
- уравнение
Леннарда-Джонса
,
c,
b,
m
= const
В случаях взаимодействия атомов с твердой поверхностью, необходимо провести суммирование всех взаимодействий.
х– расстояние до поверхности
r – радиус действия сил притяжения
dV – объем
n – число молекул поверхности
U АДС. – энергия адсорбционного взаимодействия



В случае адсорбции притяжение усиливается. И в случае при взаимодействии типа неполярное-неполярное адсорбция преимущественно локализуется в углублениях.
Электростатическое взаимодействие.
Полярный адсорбент – неполярный адсорбат
Неполярный адсорбент – полярный адсорбат
Полярный адсорбент – полярный адсорбат.
М олекулу
адсорбата представляют как диполь, а
адсорбента – как проводник, в котором
молекула адсорбата индуцирует диполь
зеркально симметрично по отношению к
данному.
олекулу
адсорбата представляют как диполь, а
адсорбента – как проводник, в котором
молекула адсорбата индуцирует диполь
зеркально симметрично по отношению к
данному.
X – расстояние до середины
При взаимодействии возникает потенциал:
 ,
,
 - дипольный момент.
- дипольный момент.
Потенциал стремится принять максимальное значение, т.е. диполи стремятся сориентироваться перпендикулярно к поверхности.
Поскольку повышение температуры способствует росту Броуновского движения, оно приводит к торможению процесса адсорбции.
В случае электростатического взаимодействия адсорбат преимущественно локализуется на выступах.
Фундаментальное адсорбционное уравнение.
В случае адсорбции происходит перераспределение компонента, а значит, изменяется химический потенциал. Процесс адсорбции можно рассматривать как переход поверхностной энергии в химическую.
Объем слоя = 0, тогда обобщенное уравнение I и II закона термодинамики:
T
= const;
(1) = (2) =>


Для двухкомпонентной системы:
,
 ,
,
 =>
=>
=>
 - адсорбционное
уравнение Гиббса
.
- адсорбционное
уравнение Гиббса
.
Для случая адсорбции
тв. тело – газ:
,
 ,
,

 - изотерма
- изотерма
 - изобара
- изобара
 - изопикна
- изопикна
 - изостера
- изостера
Изотерма, изопикна, изостера связаны друг с другом.
Т.к. адсорбция
функция




Изотерма Генри Изотерма Лангмюра



Термодинамика. Адсорбция.
Для конденсированных сред:
 ,
,
 ,
,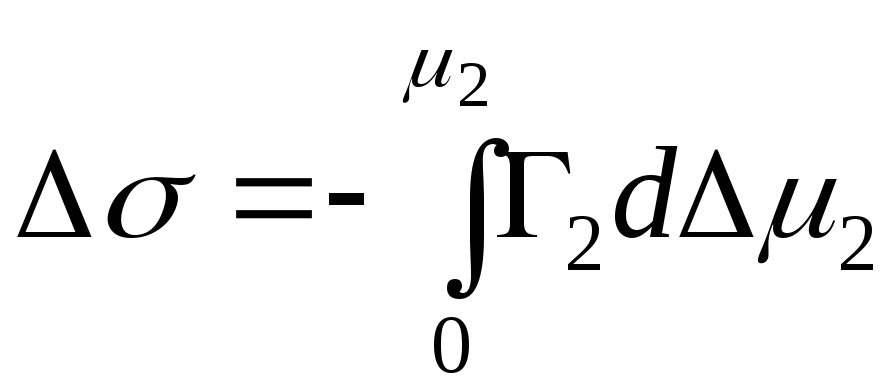
 - интегральное
изменение энергии Гиббса
.
- интегральное
изменение энергии Гиббса
.

P–давление над искривленной поверхностью, Р S –давление над плоской поверхностью
 - адсорбционный
потенциал
- адсорбционный
потенциал
Дифференциальное
изменение энтрапии

 ,
Г = const
,
Г = const
- дифференциальное изменение энтропии
- дифференциальная энтальпия адсорбции
 - изостерическая
теплота адсорбции
- изостерическая
теплота адсорбции
 - теплота
конденсации
- теплота
конденсации
 - чистая
теплота адсорбции
- чистая
теплота адсорбции

 ,
,




Qa
– интегральная теплота адсорбции,

Qra – интегральная чистая теплота адсорбции,

Уравнение Генри
Исследование адсорбции затрудняется неоднородностью поверхности, поэтому простейшие закономерности получают для однородных поверхностей.
Рассмотрим взаимодействие газов с твердой поверхностью, когда осуществляется переход газа из равновесного состояния в объеме в равновесное состояние на поверхности. Этот случай аналогичен равновесию газов в поле силы тяжести.
 ,
,
 ,
=>
,
=> -уравнение
Генри
-уравнение
Генри
 - коэффициент
распределения
- коэффициент
распределения
В процессе адсорбции происходит изменение химических потенциалов.
Для объемной фазы:

Для газа на
поверхности:

В состоянии
равновесия
 ,
т.е.
,
т.е.

В уравнении Генри константа не зависит от концентрации
Уравнение Генри выполняется в области низких давлений и концентраций. По мере роста концентрации возможны 2 типа отклонений от закона Генри:
1 – положительные отклонения, D уменьшается, А уменьшается
2 – отрицательные отклонения, D – возрастает, А – возрастает.
Тип отклонения определяется преобладанием тот или иного типа взаимодействия адсорбент-адсорбат.
При сильном адгезионном взаимодействии коэффициенты активности возрастают – положительное отклонение. В случае когезионных взаимодействий наблюдаются отрицательные отклонения.
Мономолекулрная адсорбция.
Изотерма Лангмюра.
Простейшие закономерности были получены в теории генри. Ленгмюр предложил теорию, согласно которой, адсорбция рассматривается как квазихимическая реакция. При этом:
Поверхность энергетически однородна.
Адсорбция локализована, каждый адсорбционный центр взаимодействует с одной молекулой адсорбата.
Молекулы адсорбата не взаимодействуют друг с другом.
Адсорбция монослойна.

 - поверхность,
- поверхность, - адсорбат,
- адсорбат, - адсорбционный комплекс.
- адсорбционный комплекс.
 ,
тогда концентрация адсорбционных мест:
,
тогда концентрация адсорбционных мест:
 ,
, - предельная адсорбция.
- предельная адсорбция.
 ,
тогда константа реакции:
,
тогда константа реакции:


 - уравнение Лангмюра.
- уравнение Лангмюра.
Зависимость адсорбции от концентрации
1 )
)
 ,
,
2) область высоких концентраций
 - предельная
адсорбция, образование мономолекулярного
слоя
- предельная
адсорбция, образование мономолекулярного
слоя
Для энергии Гиббса: .
g – энтропийный множитель.
В случае изотермы
Генри энергия Гиббса характеризует
переход адсорбата из стандартного
состояния в объёме в стандартное
состояние на поверхности. В случае
изотермы Ленгмюра
 характеризует степень сродства адсорбента
и адсорбата.
характеризует степень сродства адсорбента
и адсорбата.
 находят из изобары
Вант-Гоффа.
находят из изобары
Вант-Гоффа.

 ,
тогда
,
тогда
 ,
отсюда
,
отсюда .
.

 - степень заполнения
поверхности.
- степень заполнения
поверхности.




 - число свободных
мест,
- число свободных
мест,
 - число занятых мест.
- число занятых мест.
 ,
,

Т.е. в области высоких концентраций число свободных мест обратно пропорционально количеству адсорбата.
Адсорбция смеси газов на однородной поверхности.
В этом случае процесс адсорбции рассматривают как две параллельно протекающие реакции.
 (1)
(1)

 (2)
(2)


Адсорбция смеси газов на неоднородной поверхности.
В случае неоднородной поверхности нельзя ограничиваться средними заполнениями.
В следствие конкурентной борьбы, на участках различных типов возможна локализация различных адсорбатов.
В этом случае
отношение
 .
.
 ,
,
 - давление насыщенного пара адсорбата.
- давление насыщенного пара адсорбата.
 ,
,
 - теплоты адсорбции.
- теплоты адсорбции.
«+» - симбатная зависимость, «-» - антибатная зависимость, «Н» - корреляции нет.
«+» - адсорбция протекает по одинаковому механизму. На наиболее энергетически выгодных участках преимущественно адсорбируется газ, обладающий большим сродством к поверхности.
«-» - адсорбция протекает по различным механизмам и до определенного момента времени конкурентной борьбы за поверхность нет.
Мономолекулярная адсорбция преимущественно реализуется при физической адсорбции газов при малых значениях p , а также на границе раздела жидкость/газ.
Полимолекулярная адсорбция.
Теория БЭТ (Брунауэр, Эммет, Теллер).
В случае, когда образование монослоя недостаточно для компенсации поверхностной энергии, адсорбция полимолекулярна и её можно рассматривать как результат вынужденной конденсации под действием поверхностных сил.
Основные положения:
При попадании молекулы адсорбата на занятое место образуется кратный комплект.
По мере приближения p к p s уменьшается число свободных адсорбционных мест. Первоначально увеличивается, а затем уменьшается число мест, занятых единичными, двойными и т.д. комплектами.
При p =p s адсорбция переходит в конденсацию.
Горизонтальные взаимодействия отсутствуют.
Для первого слоя выполняется изотерма Лангмюра.
Поверхность рассматривается как совокупность адсорбционных мест. Справедливо условие динамического равновесия: скорость конденсации на свободных местах равна скорости испарения с занятых.
a – коэффициент конденсации (доля молекул, сконденсировавшихся на поверхности);
 ,
,
Zm – максимальное число свободных мест.
 -
частота колебаний атомов в направлении
перпендикулярном к поверхности.
-
частота колебаний атомов в направлении
перпендикулярном к поверхности.
Для первого слоя условия динамического равновесия:



 ,
тогда
,
тогда
 - уравнение Лангмюра.
- уравнение Лангмюра.
Для второго слоя
будет справедливо:

Для i-го
слоя:

Для упрощения принимают, что а и ν одинаковы для всех слоев, кроме первого. Для всех слоев кроме первого теплота адсорбции постоянна. Для последнего слоя теплота адсорбции равна теплоте конденсации. В результате было получено уравнении
 (*)
(*)
C – константа,
В случае теории БЭТ, константа С характеризует энергию Гиббса чистой адсорбции. Уравнение содержит только одну константу, а также это уравнение очень важно для определения удельной поверхности адсорбента.
Поскольку в результате адсорбции теплота выделяется, определение удельных поверхностей ведут при низких температурах.
 ????????????
????????????


Основной недостаток теории – пренебрежение горизонтальными взаимодействиями в пользу вертикальных.
Уравнение выполняется
в области значений
 от 0,05 до 0,3.
от 0,05 до 0,3.
Там, где
 <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 >
0,3 – сказывается взаимодействие адсорбат
– адсорбат.
>
0,3 – сказывается взаимодействие адсорбат
– адсорбат.
Учет взаимодействий адсорбат-адсорбат.
Взаимодействия проявляются при адсорбции на неполярной поверхности разветвленных молекул или молекул. Способных образовывать ассоциаты. В этом случае изменяется форма изотерм адсорбции.
А дсорбент
не полярен.
дсорбент
не полярен.
Графику 1 соответствуют слабые взаимодействия адсорбат-адсорбат, сильное адсорбат-адсорбент.
Графику 2 соответствуют сильное взаимодействие адсорбат-адсорбат, сильное адсорбат-адсорбент.
Графику 3 соответствуют сильное взаимодействие адсорбат-адсорбат, слабое адсорбат-адсорбент.
 ,
,

В случае взаимодействия между молекулами адсорбата необходим учет изменения коэффициентов активности. И это уравнение записывают в виде:
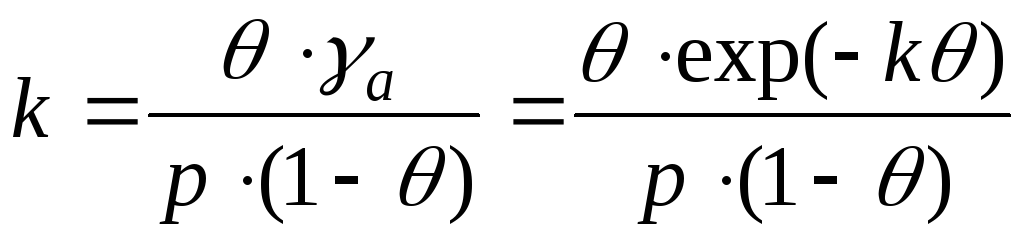 - уравнение Фрункина,
Фаулера, Гугенгейма.
- уравнение Фрункина,
Фаулера, Гугенгейма.
k – аттракционная постоянная.
Потенциальная теория Поляни.
Данная теория не выводит какого-либо типа изотермы адсорбции, а дает возможность расчета изотерм при другой температуре.
Адсорбция – это результат притяжения адсорбата к поверхности адсорбента за счет действия адсорбционного потенциала, который не зависит от присутствия других молекул и зависит от расстояния между поверхностью и молекулой адсорбата.
 ,
,
 - адсорбционный потенциал.
- адсорбционный потенциал.
Поскольку поверхность
неоднородная, расстояние заменяют на
адсорбционный объём
 .Адсорбционный
объём
– это
объём, заключенный между поверхностью
и точкой, соответствующей данному
значению
.Адсорбционный
объём
– это
объём, заключенный между поверхностью
и точкой, соответствующей данному
значению .
.
Адсорбционный потенциал – это работа перенесения 1 моль адсорбата вне данного адсорбционного объёма в данную точку адсорбционного объёма (или работа переноса 1 моль насыщенного пара адсорбата, находящегося в равновесии с жидким адсорбатом в отсутствии адсорбента в равновесную с адсорбентом паровую фазу).

Характеристическая кривая
 - адсорбционный
потенциал,
- адсорбционный
потенциал,

Для данного
адсорбента и различных адсорбатов
справедливо:

Для разных типов
адсорбатов
 ,
,
где
 потенциалы для изотерм адсорбции при
относительных давлениях
потенциалы для изотерм адсорбции при
относительных давлениях для
адсорбата 1 и для адсорбата 2. Это отношение
есть величина постоянная.
для
адсорбата 1 и для адсорбата 2. Это отношение
есть величина постоянная.
 - коэффициент
аффинности
- коэффициент
аффинности
Теория капиллярной конденсации.
Протекание процесса адсорбции во многом зависит от структуры пористого тела.
|
Микропористые | |
|
Переходнопористые | |
|
Макропористые |
В случае микропористых
сорбентов, поля адсорбционных сил
перекрываются. В случае макропористых
сорбентов, поры выполняют роль транспортных
каналов. Процессы коденсации наиболее
значимы в переходнопористых телах.
Капиллярная конденсация начинается
при определенных значениях p
и
 ,
когда часть поверхностной энергии уже
скомпенсирована. Необходимое условие
– поверхность должна быть самчиваема.
Процее описываетсяуравнением
Томпсона – Кельвина
.
,
когда часть поверхностной энергии уже
скомпенсирована. Необходимое условие
– поверхность должна быть самчиваема.
Процее описываетсяуравнением
Томпсона – Кельвина
.

 - для случая
смачивания, центр кривизны находится
в газовой фазе.
- для случая
смачивания, центр кривизны находится
в газовой фазе.
В случае капиллярной конденсации изотерма адсорбции имеет гистерезисный вид. Процессу адсорбции соответствует нижняя ветвь, процессу десорбции – верхняя.
Все виды пор можно свести к трем видам:
|
Конические |
Цилиндрические с одним закрытым концом |
Цилиндрические с двумя открытыми концами |
|
Процессное заполнение осуществляется со дна поры. Изтерма адсорбции и изотерма десорбции в этом случае совпадают, поскольку процесс адсорбции начинается со сферы и процесс десорбции также начинается с исчезновения некоторых сфер.
↓ |
Гистерезиса нет. Прямой и обратный ход описываются уравнением:
|
Дна нигде нет, заполнение поры пойдет по стенкам цилиндра.
цилиндр: Изотерма и будет иметь гистерезисный вид.
↓ |


 - сфера,
- сфера, ,
,



В условиях смачивания конденсация
осуществляется при более низких
давлениях, что энергетически выгодно.
По десорбционной ветви получают кривые
распределения пор по размерам.
условиях смачивания конденсация
осуществляется при более низких
давлениях, что энергетически выгодно.
По десорбционной ветви получают кривые
распределения пор по размерам.
Максимум
дифференциальной кривой смещен влево
относительно точки перегиба интегральной.
Общий объём малых пор невелик, однако
имеет большие значения площади
поверхности. С увеличением размера пор,
их объём возрастает как
 ,
а площадь как
,
а площадь как ,
за счет этого и наблюдается смещение
максимума дифференциальной кривой.
,
за счет этого и наблюдается смещение
максимума дифференциальной кривой.
Адсорбция на границе твердое тело – жидкость.
В случае адсорбции на границе твердое тело – газ, мы пренебрегали одним компонентом. В случае адсорбции на границе твердое тело – жидкость адсорбат вытесняет с поверхности адсорбента молекулы растворителя.
 ,
,
Справедливо уравнение:

 ,
,
N 1 , N 2 – мольные доли растворителя и компонента, N 1 + N 2 = 1, тогда
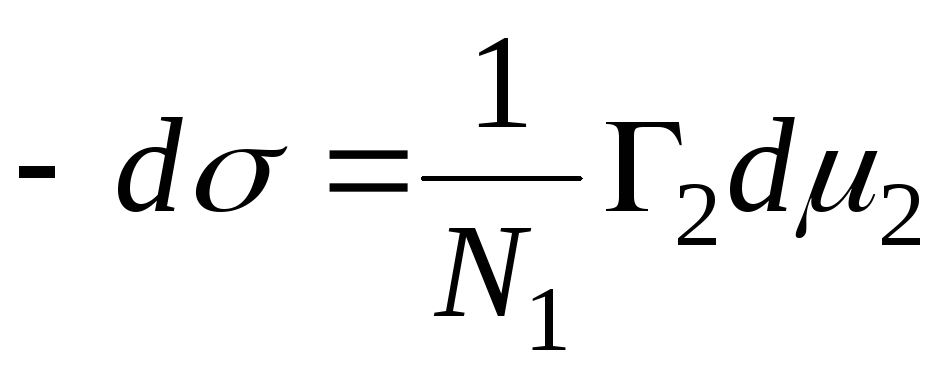 ,
=>
,
=>
 ,
тогда- уравнение адсорбции для границы раздела
фаз твердое тело – жидкость.
,
тогда- уравнение адсорбции для границы раздела
фаз твердое тело – жидкость.
Адсорбция (Г) >
0 при
 <
0
<
0
Если
значения для компонента и растворителя сильно
различны, в этом случае зависимостьГ
от N
имеет экстремум при значении N
~ 0,5.
для компонента и растворителя сильно
различны, в этом случае зависимостьГ
от N
имеет экстремум при значении N
~ 0,5.
Е сли
сли имеют
близкие значения, в этом случае возможно
изменение знака адсорбции. ЗависимостьГ
от N
пересекает ось абсцисс
имеют
близкие значения, в этом случае возможно
изменение знака адсорбции. ЗависимостьГ
от N
пересекает ось абсцисс
Точка пересечения функции Г (N ) с осью абсцисс называется адсорбционным азеотропом . Это значит, что два компонента не могут быть разделены на данном адсорбенте.
Уравнение изотермы адсорбции с константой обмена.
При адсорбции на границе твердое тело – жидкость постоянно происходит перераспределение компонентов между поверхностью адсорбента и объемом раствора.
 -
компоненты
(- - относятся к поверхности)
-
компоненты
(- - относятся к поверхности)

 ,
,
 ,
, .
.
 ,
,


Адсорбция на границе жидкость-газ
Р ассмотрим
изменение концентрационного профиля
по мере пересечения границы раздела
жидкость-газ. Пусть компонент 2 летуч.
ассмотрим
изменение концентрационного профиля
по мере пересечения границы раздела
жидкость-газ. Пусть компонент 2 летуч.
Cs – концентрация в поверхностном слое.
Исходя из определения избыточной адсорбции

Если компонент не летуч, то величина адсорбции запишется следующим образом:
П ри
ри

В уравнении
 природа вещества описывается производной
природа вещества описывается производной .
.
Изотерма поверхностного натяжения может иметь вид 1 или 2:
1 – поверхностноинактивные вещества
2 – поверхностноактивные вещества
Поверхностной активностью g называется способность веществ снижать поверхностное натяжение в системе.

 - толщина
поверхностного слоя
- толщина
поверхностного слоя
C s – концентрация компонента в поверхностном слое
С – объемная концентрация
Для гомологического ряда существует правило:
 - правило Траубо
Дюкло
- правило Траубо
Дюкло
Для гомологического ряда изотерма адсорбции выглядит таким образом:
Вместо A пишем Г, так как адсорбция избыточная в поверхностном слое.
Изотерма поверхностного натяжения:

 - поверхностное
натяжение чистого растворителя.
- поверхностное
натяжение чистого растворителя.
 - фундаментальное
адсорбционное уравнение;
- фундаментальное
адсорбционное уравнение;
 -
уравнение Лангмюра.
-
уравнение Лангмюра.
Решим их совместно:

- уравнение Шишковского.
B – константа для гомологического ряда.
A
- при переходе от одного гомолога к
другому увеличивается в 3-3,5 раза

![]()
1 – область малых концентраций
![]()
2 – средняя концентрация
3 – мономолекулярный слой
Поверхностноактивные вещества представляют собой дифильные молекулы, т.е. включают полярную группу и неполярный углеводородный радикал.
o - полярная часть молекулы.
| - неполярная часть молекулы.
В полярном растворителе молекулы ПАВ ориентируются таким образом, что полярная часть молекулы обращена к растворителю, а неполярная выталкивается в газовую фазу.
В уравнении
Шишковского
 ,
она постоянна для гомологического ряда.
,
она постоянна для гомологического ряда.
Поверхностноактивное действие начинает проявляться с n >5. При концентрациях больших, чем концентрация мономолекулярного слоя, в растворах ПАВ происходит мицеллообразоваие.
Мицелла – называется агрегат молекул дифильных ПАВ, углеводородные радикалы которых образуют ядро, а полярные группы обращены в водную фазу.
Масса мицеллы – мицелляльная масса.
Ч исло
молекул – число агрегации.
исло
молекул – число агрегации.
Сферические мицеллы
В случае мицеллообразования в растворе устанавливается равновесие
ККМ – критическая концентрация мицеллообразования.
Поскольку мы считаем мицеллу отдельной фазой:
Для гомологического ряда существует эмпирическое уравнение:
a – энергия растворения функциональной группы.
b
 – инкремент адсорбционного потенциала,
работа адсорбции на одно метиленовое
звено.
– инкремент адсорбционного потенциала,
работа адсорбции на одно метиленовое
звено.
Наличие в мицеллах углеводородного ядра создает возможность для растворения в водных растворах ПАВ соединений, которые не растворимы в воде, это явление называется солюбилизацией (то, что растворяется – солюбилизат, ПАВ – солюбилизатор).
Грязь может быть совсем не полярна, может содержать как полярную, так и не полярную часть и будет ориентироваться как молекула ПАВ.
В любом случае при солюбилизации происходит увеличение мицеллярной массы и числа агрегации не только за счет включения солюбилизата, но и за счет увеличения числа молекул ПАВ, необходимых для поддержания равновесного состояния.
Солюбилизация тем эффективнее, чем меньше молекулярная масса солюбилизата.

 ~
72 мН\м.
~
72 мН\м.
 ~
33 мН\м.
~
33 мН\м.
Эффективность ПАВ зависит от величины ККМ.
Двухмерное давление поверхностного слоя

→ -силы поверхностного натяжения.
- двухмерное давление.
Поверхностный слой – это сила равная разности поверхностных натяжений раствора ПАВ и чистого растворителя, направленная в сторону чистой поверхности.
Между раствором и поверхностным слоем устанавливается равновесие



При
 существует область, где
существует область, где линейно зависит
от концентрации.
линейно зависит
от концентрации.
Г [моль/м 2 ].
 -площадь, занимаемая
одним молем вещества
-площадь, занимаемая
одним молем вещества
Тогда изотерма
двухмерного давления будет иметь вид

 - изотерма двухмерного
давления.
- изотерма двухмерного
давления.
Зависимость
 отS М:
отS М:

При
 - двухмерное давление резко возрастает.
При
- двухмерное давление резко возрастает.
При двухмерный деформируется, что вызывает
резкий рост
двухмерный деформируется, что вызывает
резкий рост .
.
Пленка с обеих сторон ограниченная одинаковыми фазами называется двусторонней. В таких пленках наблюдается постоянное движение маточного раствора.
Пленки толщиной меньше 5 нм называются черными пленками.

Адсорбционные слои должны обладать двумя характеристиками: вязкость и легкоподвижность, текучесть и упругость.
Эффект Марангони – это самозалечивание.
Треугольник Гиббса,
 - избыточное давление.
- избыточное давление.
Пленка растянулась и за счет того, что часть жидкости ушла, ПАВ усремляются в свободное место. Треугольник Гиббса.
Эффект адсорбционной прочности тел.
На поверхности пленки всегда существует адсорбционный слой, для которого , тогда
Уравнение Лангмюра:


 в двухмерное
давление
в двухмерное
давление
 - аналог уравнения
Шишковского
- аналог уравнения
Шишковского
Электрокинетические явления. Двойной электрический слой (ДЭС).
Модель Гелемгольца. Теория Гуи-Чапмена.
1808 г. Рейс

U – образная трубка, погружают в неё 2 электрода. Закон сообщающихся сосудов нарушается и происходит изменение уровня жидкости в трубке – электрокинетические явления.
Кинетические явления:
Электрофорез
Электроосмос
Потенциал течения (протекания)
Потенциал седиментации
1 и 2 возникают при наложении разности потенциалов, 3 и 4 продавливание и седиментация коллоидных частиц вызывают появление разности потенциалов.
Электроосмосом называется движение дисперсионной среды относительно неподвижной дисперсной фазы под действием электрического тока.
Электрофорез – это движение частиц дисперсной фазы относительно неподвижной дисперсионной среды под действием электрического тока.
П ричина
возникновения электрокинетических
явлений – это пространственного
разделение зарядов и возникновение
двойного электрического слоя.
ричина
возникновения электрокинетических
явлений – это пространственного
разделение зарядов и возникновение
двойного электрического слоя.
Двойной электрический
слой представляет собой плоский
конденсатор, одна обкладка образована
потенциалоределяющими ионами, другая
– противоиноами. Ионы заражены также
как потенциалопределяющие ко-ионы
оттеснены в объем раствора. Расстояние
между обкладками
 .
Потенциал падает линейно, разность
потенциалов
.
Потенциал падает линейно, разность
потенциалов .
.
Внешняя разность
потенциалов вызывает появление модуля
сдвига
 - это пара сил отнесенных к единице
площади, девствующих вдоль поверхности
твердого тела.
- это пара сил отнесенных к единице
площади, девствующих вдоль поверхности
твердого тела.

В состоянии
равновесия модуль сдвига равен модулю
вязкого трения ( ).
).

В наших условиях
 ,
,

 - уравнение
Гелемгольца-Смалуковского
- уравнение
Гелемгольца-Смалуковского
 - линейная скорость
смещении я фаз.
- линейная скорость
смещении я фаз.
E – напряженность электрического поля.
 - разность потенциалов
между обкладками
- разность потенциалов
между обкладками
 - электрофоретическая
подвижность [м 2 /(В*с)].
- электрофоретическая
подвижность [м 2 /(В*с)].
В модели Гелемгольца не учитывается тепловое движение молекул. Реально распределение ионов в двойном слое носит более сложный характер.
Гуи и Чапман выделили следующие причины возникновения ДЭС:
Переход иона из одной фазы в другую при установлении равновесия.
Ионизация вещества твердой фазы.
Достройка поверхности ионами, присутствующими в дисперсионной среде.
Поляризация от внешнего источника тока.
Двойной электрической слой имеет размытое или диффузное строение. Ионы стремятся равномерно распределиться во всем диффузном слое.
Диффузный слой состоит из противоинонв, протяженность слоя определяется их кинетической энергией. При температуре стремящейся к абсолютному нулю противоиноы максимально приближены к потенциалопределяющим ионам.
Даня теория базируется на двух уравнениях:
уравнение Больцмана

 - работа против
сил электростатического взаимодействия.
- работа против
сил электростатического взаимодействия.
 - объёмная плотность
заряда.
- объёмная плотность
заряда.

Уравнение Пуассона

Поскольку толщина
ДЭС много меньше размеров частицы и для
плоского ДЭС производная по координатам
 и
и упраздняется.
упраздняется.

Для е у при у<<1 функцию можно разложить в ряд Маклорена:

Ограничимся двумя членами ряда, тогда:


 -
толщина ДЭС – это расстояние, на котором
потенциал ДЭС уменьшается в e
раз.
-
толщина ДЭС – это расстояние, на котором
потенциал ДЭС уменьшается в e
раз.
Чем меньше
температура, тем меньше
 .
При Т→0 – плоский ДЭС. Чем больше
концентрация, тем большеI,
тем меньше
.
При Т→0 – плоский ДЭС. Чем больше
концентрация, тем большеI,
тем меньше
 .
.





 «–» означает, что
потенциал с расстоянием уменьшается.
=>
«–» означает, что
потенциал с расстоянием уменьшается.
=>
=>

 ,
,
 - потенциал экспоненциально уменьшается.
- потенциал экспоненциально уменьшается.
Потенциал для
поверхностной плотности заряда:

Поверхностный заряд – объемный заряд с противоположным знаком, проинтегрированный по расстоянию.




=>


Там, где потенциал
уменьшается в 2,7 раза -

Ёмкость двойного
слоя

Недостаток теории
– не учитывается наличие слоя Гелемгольца,
т.е. не учитывает
 ,
отсюда ошибки в определении основных
параметров. Также не объясняет влияние
ионов различной природы на толщину
двойного электрического слоя.
,
отсюда ошибки в определении основных
параметров. Также не объясняет влияние
ионов различной природы на толщину
двойного электрического слоя.
Теория Штерна. Строение коллоидной мицеллы.
Двойной электрический
слой состоит из двух частей: плотной и
диффузной. Плотный слой образуется в
результате взаимодействия потенциалобразующих
ионов со специфически адсорбирующимися.
Эти ионы, как правило, частично или
полностью дегидратированы и могут иметь
как одинаковый, так и противоположный
к потенциалопределяющим ионам заряд.
Это зависит от соотношения энергии
электростатического взаимодействия
 и потенциала специфической адсорбции
и потенциала специфической адсорбции .
Ионы плотного слоя закреплены. Другая
часть ионов расположена в диффузном
слое, эти ионы свободны и могут перемещаться
вглубь раствора, т.е. из области большей
концентрации в область меньшей. Общая
плотность заряда складывается из двух
частей.
.
Ионы плотного слоя закреплены. Другая
часть ионов расположена в диффузном
слое, эти ионы свободны и могут перемещаться
вглубь раствора, т.е. из области большей
концентрации в область меньшей. Общая
плотность заряда складывается из двух
частей.

 -заряд слоя
Гельмгольца
-заряд слоя
Гельмгольца
 -Заряд диффузного
слоя
-Заряд диффузного
слоя
Поверхность имеет определенное число адсорбционных центров, каждый из которых взаимодействует с одним противоионом. Константа такой квазихимической реакции равна:

 ,
где
,
где
 - мольная доля противоионов в растворе
- мольная доля противоионов в растворе
Распределение Гельмгольца
Потенциал убывает линейно
Распределение
потенциала по Гуи
.
Плотного слоя нет, потенциал убывает
экспоненциально со значения

Распределение по Штерну .
Вначале снижение потенциала линейно, а затем экспоненциально.
При наложении электрического поля в случае электрофореза движется не непосредственно частица твердой фазы, а частица твердой фазы со слоем окружающих её ионов. ДЭС повторяет форму частицы дисперсной фазы. При наложении потенциала отрывается часть диффузного слоя. Линия разрыва называется границей скольжения .
Потенциал,
возникающий на границе скольжения в
результате отрыва части диффузного
слоя, называется электрокинетическим
потенциалом
(Дзэта потенциал
 ).
).
Частица дисперсной фазы, с окружающим её слоем противоионов и двойным электрическим слоем называется мицеллой .
Правила написания коллоидных мицелл:


1-1 зарядный электролит
T – частица дисперсной фазы.
AA – граница плотной и диффузной части.
BB – граница скольжения.
Граница скольжения может совпадать с линией AA, а может и не совпадать.
Значения pH, при котором дзэта-потенциал равен нулю, называется изоэлектрической точкой .
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. В избытке CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
{CaSO 4 m∙nCa 2+ 2(n - x )Cl - } 2 x + x Cl - - запись мицеллы.
CaSO 4 m – агрегат.
CaSO 4 m∙nCa 2+ – ядро.
CaSO 4 m∙nCa 2+ 2(n - x )Cl - - частица.
2. В избытке Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
{CaSO 4 m∙nSO 4 2- 2(n-x)Na + } 2x- 2xNa + - мицелла
CaSO 4 m – агрегат.
CaSO 4 m∙nSO 4 2 + – ядро.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - частица
Уравнение Гелемгольца-Смолуховского

 - линейная скорость
смещения границ (в электроосмосе).
- линейная скорость
смещения границ (в электроосмосе).
 - разность потенциалов
на обкладках конденсатора (в электроосмосе).
- разность потенциалов
на обкладках конденсатора (в электроосмосе).


 - объемная скорость
течения раствора, S
– площадь поперечного сечения ячейки.
- объемная скорость
течения раствора, S
– площадь поперечного сечения ячейки.

E – напряженность электрического поля.


 (для электроосмоса).
(для электроосмоса).
Для потенциала течения:

 - потенциал
- потенциал
 - давление на
мембрану
- давление на
мембрану
Как правило, значение электрофоретических подвижностей и электроосмотических подвижностей меньше расчетных. Это происходит вследствие:
Релаксационного эффекта (при движении частицы дисперсной фазы нарушается симметрия ионной атмосферы).
Электрофоретического торможения (возникновение дополнительного трения в результате движения противоионов).
Искажение линий тока в случае электропроводных частиц.
Связь поверхностного натяжения с потенциалом. Уравнение Липпмана.
Образование ДЭС происходит самопроизвольно вследствие стремления системы снизить свою поверхностную энергию. В условиях постоянства T и p обобщенное уравнение первого и второго законов термодинамики выглядит:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1-е уравнение
Липпмана.
- 1-е уравнение
Липпмана.
 - поверхностная
плотность заряда.
- поверхностная
плотность заряда.
 - дифференциальная
емкость.
- дифференциальная
емкость.
 - 2-е уравнение
Липпмана.
- 2-е уравнение
Липпмана.
С – емкость.
Решим 1-е уравнение Липпмана и фундаментальное уравнение адсорбции:
 ,
,


 ,
тогда
,
тогда
 - уравнение
Нернста
- уравнение
Нернста
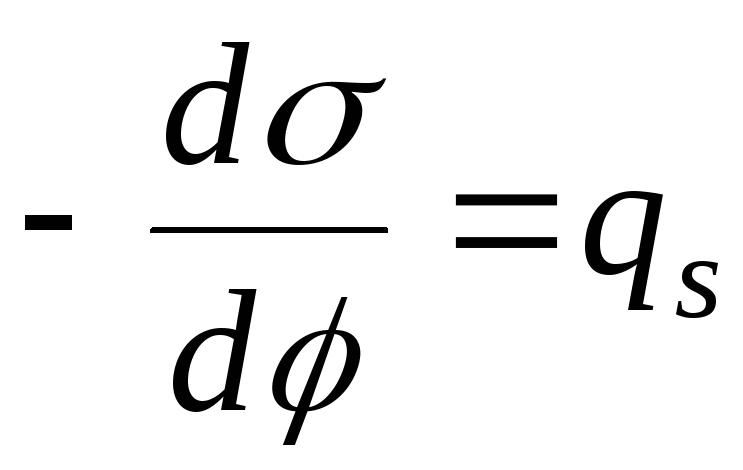 ,
,
 ,
,




 - уравнение
электрокапиллярной кривой (ЭКК).
- уравнение
электрокапиллярной кривой (ЭКК).
В
 :
: ,
но
,
но
Катионные ПАВы (КПАВ) снижают катодную ветвь ЭКК.
Анионные ПАВы (АПАВ) снижают анодную ветвь ЭКК.
Неионогенные ПАВы (НПАВ) снижают среднюю часть ЭКК.
Устойчивость дисперсных систем. Расклинивающее давление.
Дисперсные системы можно разделить:
Системы неустойчивые термодинамически могут быть устойчивы кинетически за счет перехода в метастабильное состояние.
Различают два вида устойчивости:
Седиментационная устойчивость (по отношению к силе тяжести).
Агрегативная устойчивость. (по отношению к слипанию)
Коагуляция – это процесс слипания частиц, приводящий к потере агрегативной устойчивости. Коагуляцию может вызвать изменение температуры, pH, перемешивание, ультразвук.
Различают коагуляцию:
Обратимая.
Необратимая.
Коагуляция протекает при введении электролитов.
Правила коагуляции:

Плёнка – это часть системы, находящаяся между двумя межфазными поверхностями.
Расклинивающее давление возникает при резком уменьшении толщины плёнки в результате взаимодействия сближающихся поверхностных слоев.

«-» - при уменьшении толщины пленки расклинивающее давление возрастает.
P 0 – давление в объемной фазе, которая является продолжением прослойки.
P 1 – давление в пленке.
Теория устойчивости. ДЛФО (Дерягин, Ландау, Фервей, Овербек).
Согласно теории ДЛФО в расклинивающем давлении выделяют две составляющие:
Электростатическая П Э (положительная, она обусловлена силами электростатического отталкивания). Соответствует уменьшению энергии Гиббса при возрастании толщины пленки.
Молекулярная П М (отрицательная, обусловлена действием сил притяжения). Обусловлена сжатием пленки за счет химических поверхностных сил, радиус действия сил десятые доли нм с энергией порядка 400 кДж/моль.
Полная энергия
взаимодействия
:

 - система агрегативно
устойчивая
- система агрегативно
устойчивая
 - неустойчивая
система
- неустойчивая
система
П оложительная
составляющая.
оложительная
составляющая.
Увеличение обусловлено увеличением потенциальной энергии при сжатии тонких пленок. Для пленок большой толщины избыточная энергия ионов скомпенсирована и равна энергетическому взаимодействию в объеме дисперсионной среды.
Если
 (
( - толщина пленки,
- толщина пленки, -
радиус иона) утоньшение пленки приводит
к исчезновению и уменьшению в ней молекул
и ионов с минимальной поверхностной
энергией. Число соседних частиц
уменьшается, в результате чего
потенциальная энергия оставшихся в
пленке частиц возрастает.
-
радиус иона) утоньшение пленки приводит
к исчезновению и уменьшению в ней молекул
и ионов с минимальной поверхностной
энергией. Число соседних частиц
уменьшается, в результате чего
потенциальная энергия оставшихся в
пленке частиц возрастает.


Теория ДЛФО взаимодействие частиц рассматривает как взаимодействие пластин.

Частицы не взаимодействуют



 - уравнение Лапласа,
- уравнение Лапласа,
 ,
,


Для слабо заряженных поверхностей
Для сильно заряженных поверхностей:

Молекулярная составляющая – взаимодействие двух атомов:
 ~
~
Взаимодействие атома с поверхностью:

Возьмем две пластинки:
Д ля
получения молекулярной составляющей
необходимо провести суммирование всех
энергий взаимодействия атомов правой
и левой пластин.
ля
получения молекулярной составляющей
необходимо провести суммирование всех
энергий взаимодействия атомов правой
и левой пластин.
где
 - постоянная Гамакера (учитывает природу
взаимодействующих тел).
- постоянная Гамакера (учитывает природу
взаимодействующих тел).
Т.о. энергия взаимодействия частиц в системе может быть выражена с помощью потенциальных кривых.
I – первичный потенциальный минимум. Это зона необратимой коагуляции, силы притяжения преобладают.
II – зона агрегативной устойчивости, преобладают силы отталкивания.
III – вторичный потенциальный минимум (или зона флокуляции). Между частицами дисперсной фазы существует прослойка электролита, и частицы могут быть разделены и переведены в зону агрегативной устойчивости.

Кривая 1 – система агрегативно устойчива.
Кривая 2 – в зоне I устойчива, в зоне II не устойчива.
Кривая 3 – в системе произошла коагуляция.
Кривая 4 – в точке
4 суммарная энергия взаимодействия U=0,
 ,
эта точка экстремума соответствует
началу быстрой коагуляции.
,
эта точка экстремума соответствует
началу быстрой коагуляции.
Существует два случая:
1. Поверхности слабозаряженные:
U = U Э + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - это толщина
прослойки соответствующая началу
процессу коагуляции.
- это толщина
прослойки соответствующая началу
процессу коагуляции.






 - для слабозаряженных
поверхностей
- для слабозаряженных
поверхностей
 тогда
тогда

2. Для сильнозаряженных поверхностей:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Возведем (3) в квадрат

Коагуляция:
При специфической адсорбции ионы могут адсорбироваться в сверхэквивалентном количестве таким образом, что поверхность может изменить свой заряд. Происходит перезарядка поверхности.
В случае специфической адсорбции могут адсорбироваться ионы не только противоположных знаков, но и одного.
Если адсорбируются ионы того же знака, что и поверхность, то в поверхностном слое будет происходить не падение потенциала, а его рост.

Нейтрализационная коагуляция (протекает с участием слабозаряженных частиц и зависит не только от заряда электролита-коагулятора, но и от потенциала на границе плотного и диффузного слоя).
Теория быстрой коагуляции Смолуховского.

Зависимость скорости коагуляции от концентрации электролита.
I – скорость коагуляции мала,
II – скорость коагуляции практически пропорциональна концентрации электролита.
III – область быстрой коагуляции, скорость практически не зависит от концентрации.
Основные положения :
Исходный золь монодисперсный, сходные частицы имеют сферическую форму.
Все столкновения частиц результативны.
При столкновении двух первичных частиц образуется вторичная. Вторичная + первичная = третичная. Первичное, вторичное, третичное – кратность.
В терминах химической кинетики процесс коагуляции может быть описан уравнением:

Решением будет
уравнение:

 - время половинной
коагуляции. Это время, в течение которого
число частиц золя уменьшается в 2 раза.
- время половинной
коагуляции. Это время, в течение которого
число частиц золя уменьшается в 2 раза.

 ,
,
 ,
,
 ,
,


По мере увеличения
кратности максимум кривых коагуляции
сдвигается в сторону больших значений
 .
.
Недостатки:
Предположение о монодисперсности.
Предположение о результативности всех столкновений.
Газ, не действующий химически на жидкость, может тем не менее поглощаться ею при соприкосновении с ней. Такое явление называется абсорбцией.
Для конкретности представим себе что на дне закрытого сосуда находится вода, а над водой - газообразный кислород. Некоторые молекулы кислорода будут проникать в воду и странствовать между ее молекулами. Другие кислородные молекулы будут, наоборот, вылетать из жидкости в газовую атмосферу над ней. Когда вода и кислород находятся в равновесии, то число молекул кислорода, переходящих за единицу времени из газообразной фазы в жидкую, будет равно числу молекул, переходящих за то же время из жидкой фазы в газообразную.
Если давление кислорода увеличим вдвое, то число кислородных молекул, имеющих шансы быть поглощенными жидкостью, увеличится вдвое (если поглощенное ранее количество молекул газа не так велико чтобы препятствовать дальнейшему поглощению его).
Отсюда вытекает закон установленный английским ученым Генри в 1803 г. при не слишком больших давлениях газа абсорбируемое количество газа (при данной температуре) пропорционально его давлению.
Легко сообразить, что, поскольку справедлив закон Генри, объем газа абсорбированного при данной температуре данным количеством жидкости, будет при всяком давлении выражаться одним и тем же числом Например, 1 объем воды поглощает при объем углекислого газа, 0,035 объема кислорода, 0,017 объема азота и т. д. Числа эти называют коэффициентами абсорбции.
В связи с относительно большим поглощением водой углекислоты до недавнего времени предполагали, что водяные растения дышат кислородом, который они усваивают из поглощенной водой углекислоты. Однако в 1940 г. советские ученые Виноградов и Тейсс показали, что зеленые растения в воде дышат кислородом воды, а не
Вследствие того, что коэффициент абсорбции, т. е. растворимость, кислорода в воде в два раза больше, чем коэффициент абсорбции азота, состав воздуха в воде («водяного воздуха») существенно отличается от состава атмосферного воздуха. Атмосферный воздух содержит по объему 78% азота и 21% кислорода; воздух, выделяемый из воды, содержит 63% азота и 36% кислорода. Обогащенность «водяного воздуха» кислородом имеет, по-видимому, большое биологическое значение.
Подобно тому как в системе жидкости и ее насыщенного пара повышение температуры благоприятствует переходу молекул из жидкой фазы в парообразную, так в системе жидкости и газа, ею абсорбируемого, повышение температуры благоприятствует переходу молекул газа из жидкости в газообразную фазу; это значит, что с повышением температуры коэффициент абсорбции уменьшается. Впрочем, многие металлы представляют собой исключение из этого правила.
Способностью соды абсорбировать при пониженной температуре и повышен ном давлении значительное количество углекислоты широко пользуются для изготовления шипучих напитков.
Известно, что при постепенном нагревании воды из нее выделяется все больше и больше газовых пузырьков; это - результат уменьшения коэффициента абсорбции. Кипячением можно совершенно освободить воду от абсорбированных ею газов.
Из смеси газов жидкость поглощает такое количество каждого газа, какое соответствует его парциальному давлению. Поэтому, например, количество поглощаемой углекислоты не возрастет, если в занимаемое ею над водой пространство накачать воздух.
Твердые металлы также обладают способностью поглощать газы. Так, платина, железо и другие металлы в калильном жару поглощают водород, а железо легко поглощает также окись углерода газы эти удерживаются металлами и по охлаждении последних (это явление называется окклюзией).
Строго говоря, под абсорбцией понимают только те случаи поглощения газов, когда поглощаемый газ растворяется в объеме поглощающего вещества (безразлично - жидкости или твердого тела). При поглощении газов твердыми мелкозернистыми или пористыми телами большая часть поглощенного газа не распределяется по всему объему, а удерживается в весьма уплотненном виде на поверхности пор и зерен; такое поглощение газа называют адсорбцией (§ 131). Таким образом, абсорбция - это, в сущности, растворение газа, а адсорбция - его уплотнение на микроповерхности тел. Следует отметить, однако, что при поглощении газов металлами, имеющими микрозернистое строение, явления адсорбции и абсорбции не всегда могут быть точно разграничены.

Закон Генри можно сформулировать следующим образом: при разбавлении системы (уменьшение давления) коэффициент распределения стремится к постоянному значению, равному константе распределении Генри. Относительно величины адсорбции А этот закон запишется так:
Э ти
уравнения представляют собой изотермы
адсорбции вещества при малых концентрациях.
В соответствии с ними закон Генри можно
сформулировать так: величина адсорбции
при малых давлениях газа (концентрациях
вещества в растворе) прямо пропорциональна
давлению (концентрации).
ти
уравнения представляют собой изотермы
адсорбции вещества при малых концентрациях.
В соответствии с ними закон Генри можно
сформулировать так: величина адсорбции
при малых давлениях газа (концентрациях
вещества в растворе) прямо пропорциональна
давлению (концентрации).
Отклонения от закона Генри, выражаемые изменениями коэффициентов активности в фазах, обычно не позволяют описать и прогнозировать ход изотерм с увеличением концентрации.
(давления) адсорбата. Чтобы получить теоретическую изотерму адсорбции, описывающую более широкую область концентраций, необходимо использование представлений о механизме адсорбции и конкретных моделей.
Большую долю отклонений коэффициента активности адсорбата в поверхностном слое от единицы можно учесть, используя представление об адсорбции как о квазихимической реакции между адсорбатом и адсорбционными центрами поверхности адсорбента. В этом заключается основная идея адсорбционной теории Ленгмюра. Это положение уточняется следующими допущениями:
1) адсорбция локализована (молекулы не перемещаются по поверхности) на отдельных адсорбционных центрах, каждый из которых взаимодействует только с одной молекулой адсорбата; в результате образуется мономолекулярный слой;
2) адсорбционные центры энергетически эквивалентны - поверхность адсорбента эквипотенциальна;
3) адсорбированные молекулы не взаимодействуют друг с другом.
Лиофильные дисперсные системы. Классификация и общая характеристика пав. Термодинамика и механизм мицеллообразования. Строение мицелл пав в водных и углеводородных средах. Солюбилизация.
Все дисперсные системы в зависимости от механизма процесса их образования по классификации П. А. Ребиндера подразделяют на лиофильные, которые получаются при самопроизвольном диспергировании одной из фаз (самопроизвольное образование гетерогенной свободнодисперсной системы), и лиофобные, получающиеся в результате диспергирования и конденсации с пересыщением (принудительное образование гетерогенной свободноднсперсной системы).
Если с увеличением концентрации вещества поверхностное натяжение на границе раздела фаз понижается, то такое вещество называют поверхностно-активным. Для таких веществ поверхностная активность
Наличие гидрофильной и олеофильной частей у молекул ПАВ является характерной отличительной особенностью их строения. По способности к диссоциации в водных растворах поверхностно-активные вещества делят на ионогенные и неионогенные. В свою очередь ионогенные ПАВ подразделяют на анионные, катионные и амфолитпые (амфотерные).
1) Анионные ПАВ диссоциируют в воде с образованием поверхностно-активного аниона.
2) Катионные ПАВ диссоциируют в воде с образованием поверхностно-активного катиона.
3) Амфолитные ПАВ содержат две функциональные группы, одна из которых имеет кислый, а другая основный характер, например карбоксильную и аминную группы. В зависимости от рН среды амфолитные ПАВ проявляют анионоактивные или катионоактивные свойства.
Все ПАВ относительно поведения их в воде делят на истинно растворимые и коллоидные.
Истинно растворимые ПАВ в растворе находятся в молекулярно-дисперсном состоянии вплоть до концентраций, соответствующих их насыщенным растворам и разделению системы на две сплошные фазы.
Главной отличительной особенностью коллоидных ПАВ является способность образовывать термодинамически устойчивые (лиофильные) гетерогенные дисперсные системы (ассоциативные, или мицеллярные, коллоиды). К основным свойствам коллоидных ПАВ, обусловливающим их ценные качества и широкое применение, относятся высокая поверхностная активность; способность к самопроизвольному мицеллообразованию - образованию лиофильных коллоидных растворов при концентрации ПАВ выше некоторого определенного значения, называемого критической концентрацией мицеллообразования (KKM); способность к солюбилизации - резкому увеличению растворимости веществ в растворах коллоидных ПАВ вследствне их «внедрения» внутрь мицеллы; высокая способность стабилизировать различные дисперсные системы.
При концентрациях выше KKM молекулы ПАВ собираются в мицеллы (ассоциируют) и раствор перехолит в мицеллярную (ассоциативную) коллоидную систему.
Под мицеллой ПАВ понимают ассоциат дифильных молекул, лиофильные группы которых обращены к соответствующему растворителю, а лиофобные группы соединяются друг с другом, образуя ядро мицеллы. Число молекул, составляющих мицеллу, называют числом ассоциации, а общую сумму молекулярных масс молекул в мицелле, или произведение массы мицеллы на число Авогадро, - мицеллярной массой. Определенное ориентирование дифильных молекул ПАВ в мицелле обеспечивает минимальное межфазное натяжение на границе мицелла - среда.
П ри
концентрациях ПАВ в водном растворе,
несколько превышающихKKM,
согласно представлениям Гартли образуются
сферические мицеллы (мицеллы Гартли).
Внутренняя часть мицелл Гартли состоит
из переплетающихся углеводородных
радикалов, полярные группы молекул ПАВ
обращены в водную фазу. Диаметр таких
мицелл равен удвоенной длине молекул
ПАВ. Число молекул в мицелле быстро
растет в пределах узкого интервала
концентраций, а при дальнейшем увеличении
концентрации практически не изменяется,
а увеличивается число мицелл. Сферические
мицеллы могут содержать от 20 до 100 молекул
и более.
ри
концентрациях ПАВ в водном растворе,
несколько превышающихKKM,
согласно представлениям Гартли образуются
сферические мицеллы (мицеллы Гартли).
Внутренняя часть мицелл Гартли состоит
из переплетающихся углеводородных
радикалов, полярные группы молекул ПАВ
обращены в водную фазу. Диаметр таких
мицелл равен удвоенной длине молекул
ПАВ. Число молекул в мицелле быстро
растет в пределах узкого интервала
концентраций, а при дальнейшем увеличении
концентрации практически не изменяется,
а увеличивается число мицелл. Сферические
мицеллы могут содержать от 20 до 100 молекул
и более.
При увеличении концентрации ПАВ мицеллярная система проходит ряд равновесных состояний, различающихся по числам ассоциации, размерам и форме мицелл. При достижении определенной концентрации сферические мицеллы начинают взаимодействовать между собой, что способствует их деформации. Мицеллы стремятся принять цилиндрическую, дискообразную, палочкообразную, пластинчатую форму.
Мицеллообразование в неводных средах, как правило, является результатом действия сил притяжения между полярными группами ПАВ и взаимодействия углеводородных радикалов с молекулами растворителя. Образующиеся мицеллы обращенного вида содержат внутри негидратированные или гидратированные полярные группы, окруженные слоем из углеводородных радикалов. Число ассоциации (от 3 до 40) значительно меньше, чем для водных растворов ПАВ. Как правило, оно растет с увеличением углеводородного радикала до определенного предела.
Явление растворения веществ в мицеллах ПАВ называется солюбилизацией. Способ включения молекул солюбилизата в мицеллы в водных растворах зависит от природы вещества. Неполярные углеводороды, внедряясь в мицеллы, располагаются в углеводородных ядрах мицелл. Полярные органические вещества (спирты, амины, кислоты) встраиваются в мицеллу между молекулами ПАВ так, чтобы их полярные группы были обращены к воде, а липофильные части молекул ориентированы параллельно углеводородным радикалам ПАВ. Возможен и третий способ включения солюбилизата в мицеллы, особенно характерный для неионогенных ПАВ. Молекулы солюбилизата, например фенола, не проникают внутрь мицелл, а закрепляются на их поверхности, располагаясь между беспорядочно изогнутыми полиоксиэтиленовыми цепями.
Солюбилизация - самопроизвольный и обратимый процесс; данной концентрации ПАВ и температуре соответствует вполне определенное насыщение раствора солюбилизатом. В результате солюбилизации получаются устойчивые дисперсные системы подобные самопроизвольно образующимся ультрамнкрогетерогенным эмульсиям.
Определите поверхностную и полную (внутреннюю) энергию 4 г водяного тумана, имеющего частицы с дисперсностью 5·10 7 м -1 , t = 20º C , σ = 72 мДж/м 2 ; d σ/ dT = ‑ 0,16 мДж/(м 2 ·К); ρ = 1000 кг/м 3 .

Экзаменационный билет № 10
Теopия полимолекулярной адсорбции БЭТ: исходные положения, вывод уравнения изотермы и его анализ. Линейная форма уравнения БЭТ. Определение удельной поверхности адсорбентов, катализаторов и других пористых тел.
Уравнение Ленгмюра можно использовагь только при условии, что адсорбция вещества сопровождается образованием мономолекулярного слоя.
В большинстве случаев мономолекулярный адсорбционный слой не компенсирует полностью избыточную поверхностную энергию и влияние поверхностных сил может распространяться на второй, третий и последующие адсорбционные слои, в результате проходит полимолекулярная адсорбция.
С овременная
форма уравнения полимолекулярной
адсорбции - основного уравнения
обобщенной теории Ленгмюра - была
предложена Брунауэром, Эмметом и
Теллером.
овременная
форма уравнения полимолекулярной
адсорбции - основного уравнения
обобщенной теории Ленгмюра - была
предложена Брунауэром, Эмметом и
Теллером.
В этой теории дополнительным допущением к тем, которые были положены в основу вывода уравнения изотермы Ленгмюра, является представление об образовании на поверхности адсорбента «последовательных комплексов» адсорбционных центров с одной, двумя, тремя и т. д. молекулами адсорбата. Тогда процесс адсорбции можно представить в виде последовательных квазихимических реакций:
Константы равновесия этих реакций соответственно равны
Обозначим:
Общее число активных центров на адсорбенте, или емкость монослоя, будет равна

После ряда вычислений с применением теории рядов, окончательно получим:

Д анное
соотношение является основным уравнением
обобщенной теории Ленгмюра и называется
уравнением полимолекулярной адсорбции
БЭТ.
анное
соотношение является основным уравнением
обобщенной теории Ленгмюра и называется
уравнением полимолекулярной адсорбции
БЭТ.
При обработке экспериментальных результатов уравнение БЭТ обычно используют в линейной форме:
Оно позволяет графически определить оба постоянных параметра A ∞ и С:
Экспериментальное
определение A ∞
позволяет рассчитать удельную поверхность
адсорбента (поверхность единицы массы
адсорбента): ![]() .
.